What is phylogenetics?
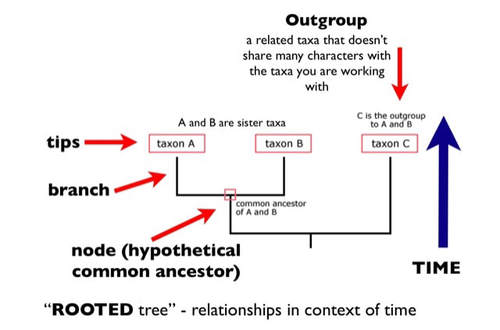
Figure 1. Phylogeny Tree Diagram
Phylogenetics is the study of evolutionary relationships among biological entities, such as species, individuals, or genes - which are often referred to as taxa. [1] Phylogenetic trees are constructed by looking at nucleotide or protein sequences and combining those with the understanding of sequence evolution. This allows us to infer evolutionary events that have taken place which provides information about evolutionary processes operating on sequences. [1] These trees can be used to infer ancestral linkages between different species.
There are various parts that make up phylogenetic trees. Taxa are represented by the tips of the tree branches. [2] Taxa can be at any taxonomic level such as species or populations. The lines found within the tree are called the branches. The points where branches connect are called nodes. Internal nodes connect branches while external nodes are the tips that represent taxa. [2] Some trees have a basal node which is known as the root. A grouping of an ancestor and all of its descendants is known as a clade.
There are various parts that make up phylogenetic trees. Taxa are represented by the tips of the tree branches. [2] Taxa can be at any taxonomic level such as species or populations. The lines found within the tree are called the branches. The points where branches connect are called nodes. Internal nodes connect branches while external nodes are the tips that represent taxa. [2] Some trees have a basal node which is known as the root. A grouping of an ancestor and all of its descendants is known as a clade.
Methods of Phylogenetic Inference
There are three methods that can be applied when generating and analyzing phylogenetic trees: Maximum Likelihood, Neighbor Joining and Average Distance. These three methods are different because while Neighbor Joining and Average Distance are more focused on distance-based methods to analyze pair-wise differences among the sequences, Maximum Likelihood is a character-based method. [3] Average distance trees differ from Neighbor Joining trees because they do not have brunches of different lengths.
Maximum Likelihood involves the modification of branch lengths. For this method, an initial tree is generated using a quick, efficient method such as Neighbor Joining. Once the tree is generated, the branch lengths are modified to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
Neighbor Joining involves the use of similarity scores deriving from percent identity or BLOSSUM, a bioinformatics substitution matrix used for the sequence alignment of proteins. These scores are used to determine which species are more closely related to one another. Branch lengths are calculated using these scores when generating the tree.
Average Distance involves using similarity scores to determine which species are most closely related and displays these relationships through the usage of equal branch lengths. This method works under the assumption that both species diverged equally from the common ancestor.
Maximum Likelihood involves the modification of branch lengths. For this method, an initial tree is generated using a quick, efficient method such as Neighbor Joining. Once the tree is generated, the branch lengths are modified to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
Neighbor Joining involves the use of similarity scores deriving from percent identity or BLOSSUM, a bioinformatics substitution matrix used for the sequence alignment of proteins. These scores are used to determine which species are more closely related to one another. Branch lengths are calculated using these scores when generating the tree.
Average Distance involves using similarity scores to determine which species are most closely related and displays these relationships through the usage of equal branch lengths. This method works under the assumption that both species diverged equally from the common ancestor.
Phylogenetic Tree Construction (for TCF4)
1) Obtain protein sequences from different organisms of interest.
Put these sequences into annotated FASTA format within a .txt file.
Put these sequences into annotated FASTA format within a .txt file.
2) Align these sequences up with each other using ClustalWOmega.
3) Now that the sequences are aligned, they can be constructed into a tree:
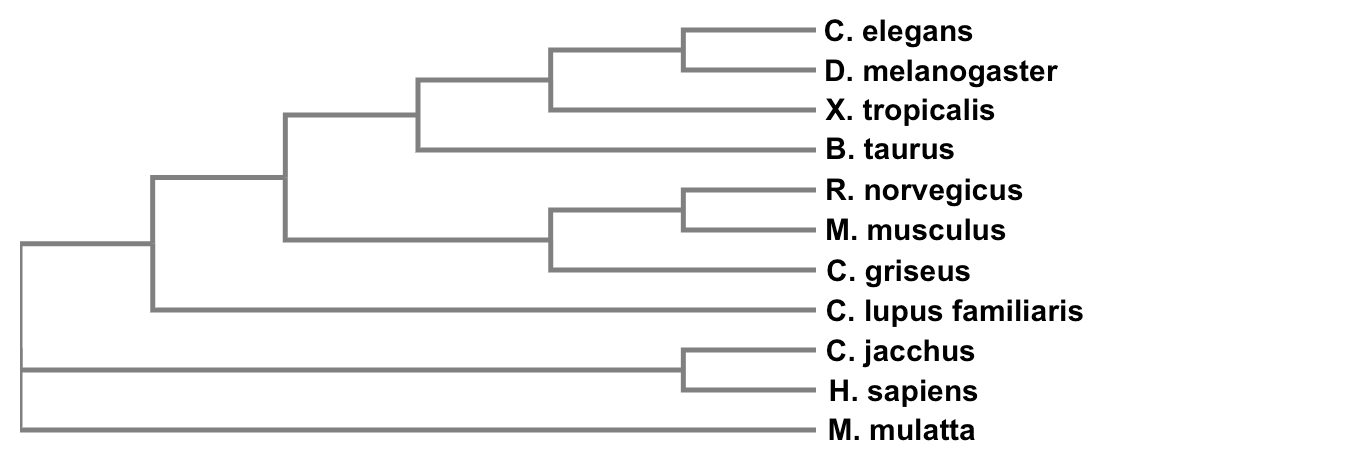
Neighbor Joining Tree
4) Make phylogenetic inferences based off of the evolutionary relationships displayed on the phylogenetic tree while using analysis methods such as maximum likelihood, neighbor joining, and average distance
Conclusion
The use of protein phylogenetics allows us to analyze the presence of TCF4 throughout several different species. In addition, it allows us to evaluate how accurately the percent identities found through BLAST reflect the evolutionary patterns shown in the phylogenetic tree that was generated. Although the percent identities do not always necessarily reflect the evolutionary patterns of a particular gene, the phylogenetic tree generated for TCF4 reflects the percent identities found via reciprocal BLASTs. The percent identities were lowest for Caenorhabditis elegans and Drosophila melanogaster, which are the two species displayed farthest from the root of the tree whereas all the other species had generally high percent identities. Therefore, it is informative to compare how the TCF4 gene is conserved among different species as well as its evolutionary patterns via protein homology or protein phylogenetics.
References
Header: http://www.zo.utexas.edu/faculty/antisense/downloadfilesToL.html
[1] EMBL-EBI. (n.d.). What is Phylogenetics? <https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics>
[2] NCBI. (2003, July 28). "Tree" Facts: Terminology. <https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo7.html>
[3] Chhotwani, M., Francis, S., & Pal, A. (n.d.). Comparison of different phylogenetic tree generation methods. Retrieved from https://cise.ufl.edu/~sarath/BioInformatics/
Figure 1. http://bcrc.bio.umass.edu/intro/manual/index.php?title=Studying_Evolutionary_Relationships&oldid=2437
Header: http://www.zo.utexas.edu/faculty/antisense/downloadfilesToL.html
[1] EMBL-EBI. (n.d.). What is Phylogenetics? <https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics>
[2] NCBI. (2003, July 28). "Tree" Facts: Terminology. <https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo7.html>
[3] Chhotwani, M., Francis, S., & Pal, A. (n.d.). Comparison of different phylogenetic tree generation methods. Retrieved from https://cise.ufl.edu/~sarath/BioInformatics/
Figure 1. http://bcrc.bio.umass.edu/intro/manual/index.php?title=Studying_Evolutionary_Relationships&oldid=2437
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
